

Tysabri (natalizumab) for multiple sclerosis
Last updated Nov. 3, 2025, by Marisa Wexler, MS
 Fact-checked by Inês Martins, PhD
Fact-checked by Inês Martins, PhD
What is Tysabri for MS?
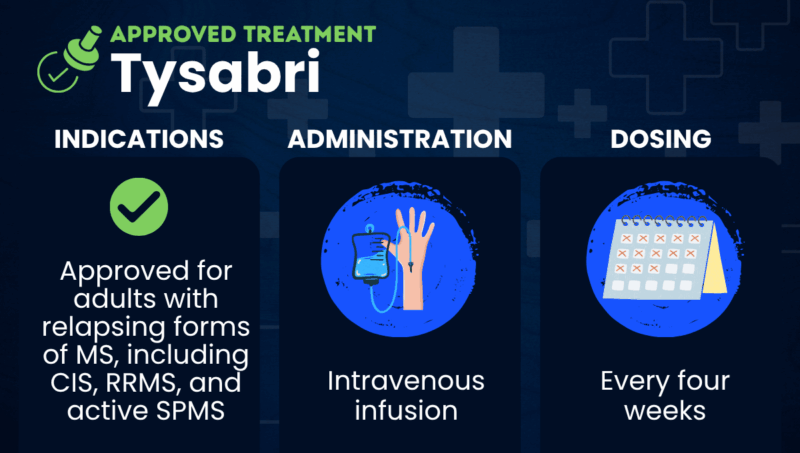
Tysabri (natalizumab) is an infusion therapy approved for adults with relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), and active secondary progressive MS (SPMS).
Administered every four weeks as an intravenous infusion, or into the bloodstream, it is used to reduce relapse rates, slow the development of brain lesions, and delay disability progression.
Tysabri works by targeting alpha-4-beta-1 integrin, a protein on immune cells that helps them cross from the bloodstream into the brain. By blocking its interaction with VCAM-1, a protein on blood vessel walls, the therapy helps prevent circulating immune cells from reaching the brain and causing damage.
Tysabri is marketed by Biogen and is also approved to treat Crohn’s disease, an inflammatory bowel disorder.
Therapy Snapshot
Scroll horizontally to view all columns -->
| Brand Name: | Tysabri |
| Chemical Name: | Natalizumab |
| Usage: | Used to reduce disease activity and delay disability progression in relapsing forms of MS |
| Administration: | Intravenous infusion |
Who with MS can take Tysabri?
Tysabri is approved in the U.S. as a standalone treatment for adults with CIS, RRMS, and active SPMS.
However, the medication’s prescribing information carries a boxed warning noting that it can increase the risk of progressive multifocal leukoencephalopathy (PML), a rare brain infection that can be fatal or lead to severe disability.
To ensure safe use, the therapy is available in the U.S. only through a restricted program that ensures healthcare professionals, pharmacies, and infusion centers are all certified to prescribe, dispense, and infuse Tysabri. Patients should be enrolled in the program and know about the risk of PML before receiving the medication.
In the U.S., Tysabri is contraindicated for anyone with:
- a current or previous PML infection
- a history of hypersensitivity reactions — a type of exaggerated immune responses, including anaphylaxis — to Tysabri.
Tysabri is approved in more than 80 countries worldwide, but the exact indications may vary. In some countries, including the European Union and Canada, it is approved only for people with RRMS.
How is Tysabri administered?
Tysabri is given via intravenous infusions that are administered by a certified professional. The recommended dose is 300 mg, which is given over about an hour every four weeks.
Tysabri in MS clinical trials
Tysabri’s approval in the U.S. was mainly based on data from two Phase 3 clinical trials:
- The AFFIRM trial (NCT00027300) tested Tysabri against a placebo in 942 adults with relapsing forms of MS for more than two years. Results showed Tysabri significantly reduced the risk of sustained disability progression by 42% and lowered relapse rates by 67% compared with the placebo. The number of new or enlarging disease lesions and lesions with active inflammation were also significantly reduced with Tysabri.
- The SENTINEL study (NCT00030966) enrolled 1,171 adults with RRMS who were given Tysabri or a placebo for more than two years while also receiving treatment with Avonex (interferon beta-1a), an approved MS therapy. Results showed that adding Tysabri to Avonex significantly reduced the risk of sustained disability progression, by 24%, compared with Avonex plus a placebo. Relapse rates were 56% lower in patients given Tysabri, and there was also a significant reduction in the number of new or enlarging lesions visible on MRI scans.
Tysabri was also tested in a long-term observational study (NCT00493298), where it was shown to be effective for more than 15 years. Among people who had at least one relapse in the year before starting Tysabri, the average number of relapses dropped by 92% in the years after the start of treatment. No new safety issues were observed with long-term treatment.
Tysabri side effects
The most common side effects of Tysabri reported in people with MS include:
- headache
- fatigue
- pain in the joints and extremities
- infections of the urinary and respiratory tracts
- gastroenteritis (inflammation of the stomach and intestines)
- vaginitis (inflammation of the vagina)
- depression
- abdominal discomfort
- diarrhea
- rash.
Tysabri carries a boxed warning that it can cause PML, an opportunistic brain infection that often is life-threatening or leads to severe disability. Patients should be monitored for this complication while on treatment, and Tysabri should be immediately stopped at the first sign of PML.
Other side effects associated with Tysabri, which may be serious, include:
- other infections, some of which can be fatal
- liver damage
- allergic reactions
- low blood cell counts, including in newborns exposed to Tysabri during pregnancy.
Patients taking Tysabri will be monitored for these issues before and throughout treatment, and should be advised on any signs and symptoms to look out for. In some cases, treatment may need to be paused or discontinued.
Multiple Sclerosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
FAQs about Tysabri in MS
Tysabri was approved by the U.S. Food and Drug Administration in November 2004 to treat relapsing forms of multiple sclerosis (MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
In the AFFIRM trial, which tested Tysabri in more than 900 people with relapsing forms of multiple sclerosis, the therapy began to demonstrably reduce the disease activity as soon as two months after starting treatment. However, as every person is unique, patients are advised to discuss with their healthcare team how the medication may help in their specific case.
While it is not yet known if Tysabri can cause birth defects, there have been reports of low blood cell and platelet counts in infants who were exposed to Tysabri during pregnancy. Those who become pregnant or plan to while on Tysabri should discuss the potential benefits and risks with their healthcare providers as early as possible.
Tysabri is not known to interact with alcohol. However, both Tysabri and alcohol can affect the liver, so drinking while on treatment may increase the risk of liver damage. Alcohol may also worsen certain multiple sclerosis symptoms, so patients should always discuss safe alcohol consumption with their healthcare provider.
Hair loss has not been reported as a side effect of Tysabri in multiple sclerosis clinical trials, but some people have experienced an increase in body weight. There are also reports of patients experiencing a decrease in body weight. Patients who experience unanticipated effects after starting treatment should talk to their healthcare team.


